数値シミュレーションは、修正されたセルオートマトンモデルのモンテカルロ実装に基づいている。模擬された結晶表面は、2次元マトリックスによって表され、ここで、元素の値および位置は、結晶の表面上の分子の3D位置を規定する。
分子を受容または放出する相対確率は、結晶成長のモデルに表面エネルギーを直接導入することができる、表面上の各分子の周囲に依存する熱活性化反応のモデルの基底で計算される。
モデルはまた、粗い表面上にボトルネックのような構造を詰めた結果形成される濃度ホールタイプの欠陥の計算を可能にする。
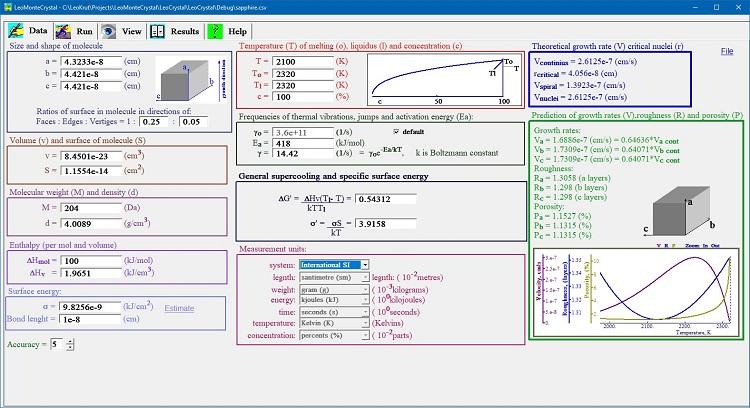
単一実験のパラメータは、データパネルに入力することができ、好きな測定単位を柔軟に選択できます。
LeoMonteCrystalは単位変換機能を提供し、ユーザーの慣習に合わせてさまざまな単位システムで定義された初期パラメータを使用できます。
データ入力パネルは、競合理論モデルで計算された速度結晶成長の理論値を表示し、与えられた初期パラメータのセットについて我々の研究の結果として見いだされた式で最も価値があります。
我々の公式によって計算された成長速度、表面粗さおよび欠陥濃度の温度依存性は、別々のチャートで表示されている。
いずれか1つのパラメータの修正後、すべての信頼できる変数が自動的に更新され、それに応じて熱力学および結晶学計算機の機能が提供される。
関心のある化合物に対応する頻繁に使用されるパラメーターのセットは、昏睡区切りファイル(MS Excel互換)との間で保存またはインポートすることができます。
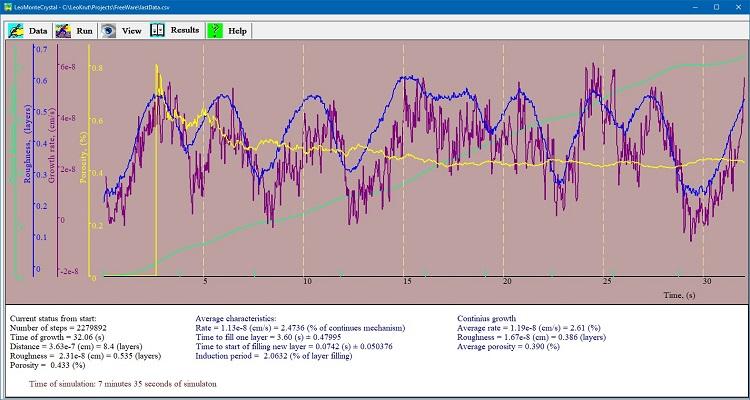
シミュレーション中の「結果」パネルには、結晶表面成長の進行距離、その粗さ、瞬間成長率、および欠陥のような穴の濃度のタイムラインを表示するチャートがあります。
このリリースの新機能:
バージョン1.1:独自の最新式で計算された成長速度、表面粗さおよび欠陥濃度の温度依存性は別々のチャートで表示されます。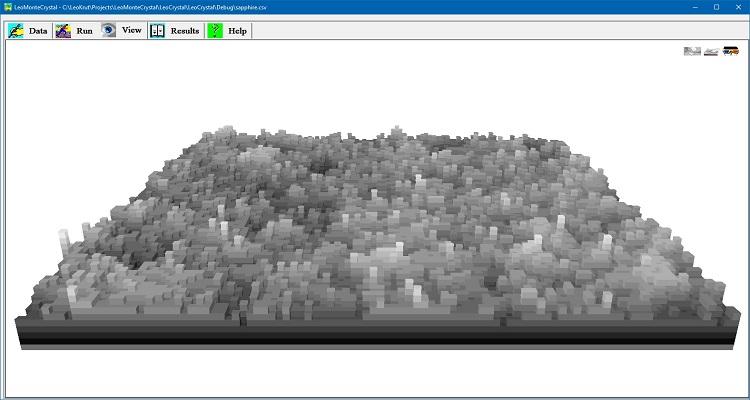

コメントが見つかりません